Tanvex Presents Positive Findings for Trastuzumab Candidate at ESMO
A phase 3 study of a trastuzumab biosimilar candidate (TX05) and reference product (Herceptin) has demonstrated equivalence in patients with human epidermal growth factor receptor 2 (HER2)-positive early stage breast cancer, according to findings presented at the European Society for Medical Oncology Congress 2021.
The trial enrolled patients (N = 809) in the neoadjuvant, or presurgery, setting. Of those, 674 were evaluable. The primary end point was therapeutic equivalence based on pathologic complete response (pCR), or absence of residual cancer, following neoadjuvant chemotherapy.
Secondary end points were objective response rate (ORR), immunogenicity, safety, and tolerability.
Patients were recruited at 124 treatment centers in 10 countries, including Russia, Hungary, Chile, and India. They received four 3-week cycles of epirubicin and cyclophosphamide and were then randomized to four 3-week cycles of paclitaxel and TX05 or the reference biosimilar (TRA, as available in the European Union). Investigators said close to 100% of patients completed all planned cycles of trastuzumab therapy.
They said the proportions of patients meeting pCR criteria for equivalence were ?highly similar? between the TX05 (48.8%) and TRA (45.3%) cohorts (risk ratio, 1.0783).
They also said equivalence was observed when patients from both cohorts were stratified according to cancer stage and Eastern Cooperative Oncology Group (ECOG) performance status. ECOG is a means of classifying patients according to the level of disability they suffer as a result of their cancer, with zero representing the highest functionality.
ORR was also highly similar between the TX05 (84.3%) and TRA (85.0%) groups, and findings for patients with complete response, partial response, and stable disease were also highly similar between cohorts. The same was true for pharmacodynamics, authors of the study said.
?In all sensitivity analyses, including a 'tipping point' analysis, the 95% CI of the risk ratio was completely contained within the predefined interval,? they wrote.
Safety analysis of treatment-emergent adverse events (TEAEs) was confined to cycles 5 through 8 of the treatment, as this was the period during which patients received trastuzumab. The investigators said 62.4% of patients experienced TEAEs with TX05 vs 62.5% of patients with TRA, which they said was similar and demonstrative of biosimilarity for TX05.
The most common TEAEs were musculoskeletal and connective tissue disorders, nervous system and gastrointestinal disorders, general disorders, and investigative-related and administration-site conditions.
?Overall, the TX05 study treatment was well tolerated. The safety profile was consistent with the known profile of TRA with no significant safety findings seen in the study,? the authors wrote.
?The results of this study support that there is no clinically meaningful difference between TX05 and [TRA],? they said, adding that based on pharmacokinetic similarity, it was possible to conclude that TX05 is also highly similar to the US version of reference trastuzumab.
The study was sponsored by Tanvex BioPharma USA, which is the company developing TX05 for market.
Preliminary results from the phase 3 study concerning just pCR were released in February 2021.
Reference
Krivorotko P, Manikhas A, Moiseenko F, et al. Trial comparing the safety, efficacy, and immunogenicity of trastuzumab biosimilar candidate (TX05) with originator trastuzumab in HER2+ early breast cancer. Presented at ESMO Congress 2021; September 17-21, 2021. ePoster 127P.
The court battle between AbbVie and Alvotech could be a defining victory in the struggle to bring biosimilar versions of adalimumab to market, but a yearlong court process must play out first.
A high-concentration, citrate-free biosimilar of adalimumab (AVT02) proposed by Alvotech of Reykjavik, Iceland, would not come to market before October 2022, a year from now, under a court agreement.
Facing litigation from AbbVie, the Lake Bluff, Illinois-based maker of the original adalimumab (Humira), Alvotech has sought to reduce the number of Humira patents it has to fight to get its biosimilar candidate to market in the United States.
In 2020, AbbVie had annual revenues approaching $20 billion for Humira, an often life-changing product approved for chronic inflammatory diseases such as rheumatoid arthritis, Crohn disease, and psoriasis.
AbbVie originally ?asserted? or claimed potential infringement of 62 patents related to adalimumab, and Alvotech countered that just?4 adalimumab patents?are of substance in the dispute over AVT02.
The District Court for the Northern District of Illinois has denied a motion by Alvotech to have the court venue changed to the Eastern District of Virginia, where the company thought it could get a speedier, more favorable decision.
The court has also whittled down the number of adalimumab patents that will be litigated in this round to 10, vs the 62 that AbbVie asserted and the 4 that Alvotech has contended are appropriate to address.
A scheduling order issued by the court in September 2021 sets forth the procedure for getting to a decision in the case by October 2022. This allows for hearings on the parties? claims and counter claims about infringement and enforceability of the adalimumab patent portfolio, a discovery process that extends through June 2022, followed by a trial set for most of August 2022.
According to the court document, AbbVie will restrict its case to the 10 patents identified by the court and will await the court?s decision before requesting an injunction to prevent Alvotech from making use of any patented intellectual property related to adalimumab.
The scheduling document also states that Alvotech has ?agreed not to launch AVT02 in the United States prior to the issuance of the court?s decision.?
There are multiple biosimilars of adalimumab already approved, although these are lower-concentration products, and settlements between AbbVie and the respective biosimilar companies will prevent any of these biosimilars from coming to market before 2023.
Alvotech proposes to bring a 100 mg/mL, citrate-free version of adalimumab to market to capitalize on the growing market dominance of this concentration that AbbVie enjoys almost exclusively.
Adalimumab can cause a severe, burning sensation upon administration, which patients may have to endure with multiple monthly injections. Citrate is used to stabilize the pH of adalimumab, and the citrate-free version uses a different buffer that doesn?t cause the same burning and pain that patients experience. This can make citrate-free adalimumab much more popular.
Adalimumab biosimilars have encountered fewer market barriers in the European Union, and during the third quarter of 2021, Celltrion Healthcare aimed to launch its?own high concentration adalimumab?product (Yuflyma) in 7 EU countries.
The company planned to launch Yuflyma in 3 additional EU countries in the fourth quarter of this year. Celltrion has yet to announce a US launch date, and, indeed, has not yet obtained FDA approval for this agent.
Alvotech is seeking an?interchangeable designation?for AVT02 which it believes would increase the use of the product in the United States. Interchangeability would allow pharmacists to substitute the biosimilar for Humira without consulting the prescribing physician.
Related Development
The US House Judiciary Committee has approved a bill (HR 2884) that would limit the number of patents an originator company could ?assert,? or use as grounds for litigation, against a biosimilar developer.
The limits would be contingent on the biosimilar company?s compliance with information exchanges recommended under the biosimilar approval pathway outlined under the Biologics Price Competition and Innovation Act.
The bill, which has yet to pass the full House or the Senate, would limit the number of patents that could be asserted to 20. An originator?s ability to file suit over patents that were awarded after the date of reference product approval also would be restricted.
The legislation is intended to address the problem of patent ?thickets? that represent formidable barriers to companies that want to bring biosimilars to market. Originator companies may have extensive patent portfolios. AbbVie has as many as 136 patents on its adalimumab product which have forced rival companies to settle for delayed market entry pacts with AbbVie rather than risk years of costly litigation.
A?recent study?by the Biotechnology Innovation Organization (BIO) found that the average number of patents asserted in biosimilar cases is 12, which is well below the 20-patent limit that HR 2884 would impose. However, the number of patents AbbVie has attempted to assert in its case against Alvotech?s proposed adalimumab biosimilar AVT02 vastly exceeds that number.
conference |?Scientific Meeting of the Retina Society
Positive findings for Coherus BioSciences' ranibizumab candidate (CHS-201) were presented at the Scientific Meeting of the Retina Society, and the company said equivalence end points were met for an on-body injector version of Udenyca (pegfilgrastim).
Coherus BioSciences has reached clinical milestones for 2 of its proposed biosimilar products, an on-body injector (OBI) kit for pegfilgrastim and a biosimilar version of ranibizumab, referencing Lucentis.
The company, based in Redwood City, California, is developing a wearable injector kit for its pegfilgrastim biosimilar (Udenyca) that would compete with Amgen?s commercially successful Onpro device, which holds a?51% share?of the market for pegfilgrastim and has enjoyed a tailwind from the COVID-19 pandemic, as it has answered the need for injections delivered outside the clinic.
However, the Onpro's share of the market for pegfilgrastim has declined overall from around 62% of the market in the third quarter of 2018. There are no other OBI pegfilgrastim products on the market aside from Neulasta Onpro.
Coherus BioSciences' pegfilgrastim biosimilar
Pegfilgrastim is used to regenerate white blood cells (neutrophils) following chemotherapy, thereby helping to prevent infection resulting from neutropenia (decreased white blood cell count).
Coherus stated also that positive findings demonstrating safety, efficacy, and immunogenicity equivalence were presented recently for the company?s ranibizumab biosimilar candidate (CHS-201), for the treatment of neovascular age-related macular degeneration (wet AMD). The findings for CHS-201 were presented October 1, 2021, at the 54th Annual Scientific Meeting of the Retina Society.
Investigators in the pivotal COLUMBUS-AMD study sought to determine CHS-201/Lucentis biosimilarity in terms of clinical efficacy, safety, and immunogenicity in patients with newly diagnosed subfoveal wet AMD.
"These findings reinforce our confidence that CHS-201 delivers outcomes and a safety profile similar to the reference product,? stated Peter K. Kaiser, MD, a professor of ophthalmology at the Cole Eye Institute of Cleveland Clinic.
?Neovascular age-related macular degeneration destroys the sharp, central vision needed to see clearly and can affect daily activities like reading, driving, and watching television. It is responsible for more than 90% of AMD-related severe visual loss, which has a significant deleterious impact on a patient?s quality of life,? he said.
Investigators randomized patients (N = 477) equally to CHS-201 or reference product every 4 weeks for up to 48 weeks. The primary end point was the improvement after 8 weeks in best corrected visual acuity (BCVA), as measured by Early Treatment Diabetic Retinopathy Study (ETDRS) letters. Secondary end points were BCVA change at 48 weeks and the change in foveal center point (FCP) retinal thickness at 48 weeks.
Patients in the study demonstrated a mean BCVA improvement from baseline at 8 weeks with an equal median improvement of 5 ETDRS letters for CHS-201 (mean [SD], 5.1 [7.52) and the reference product (mean, 5.6 [8.63]) cohorts. Investigators said the BCVA findings for CHS-201 were well within the equivalency margins established for demonstrating biosimilarity.
?Patients in both treatment groups experienced similar reductions in FCP and foveal central subfield retinal thickness, as well as total lesion area,? the investigators said.
They added that proportions of patients from both treatment groups with reduced choroidal neovascularization leakage and increased fluid-free macula also were similar.
The frequency of ocular adverse events (AEs), including intraocular inflammation, were comparable between cohorts, and most AEs were of mild or moderate intensity. The investigators said no clinically meaningful differences were identified. Immunogenicity profiles also were comparable between CHS-201 and the reference product.
On October 1, 2021, the FDA accepted Coherus? biologics license application for CHS-201 and set a decision date of August 2, 2022. The company said that if approved on schedule, the product would be launched in the second half of 2022.
There is 1?FDA-approved ranibizumab biosimilar?so far. Byooviz, a Samsung Bioepis and Biogen product, was approved in September 2021, although a commercial launch is not anticipated before June 2022.
Udenyca OBI
In the trial of the Udenyca OBI, investigators randomized patients (N = 189) equally to OBI followed by prefilled syringe or the reverse.
The company said all pharmacokinetic (PK) bioequivalence primary end points were met, as was the key secondary PK end point of absolute neutrophil count. Investigators said no new safety signals were observed. A 2022 FDA application for marketing approval is planned.
Although the company is seeking FDA approval to market the pegfilgrastim OBI, the Udenyca prefilled syringe was approved in 2018 and launched in January 2019.
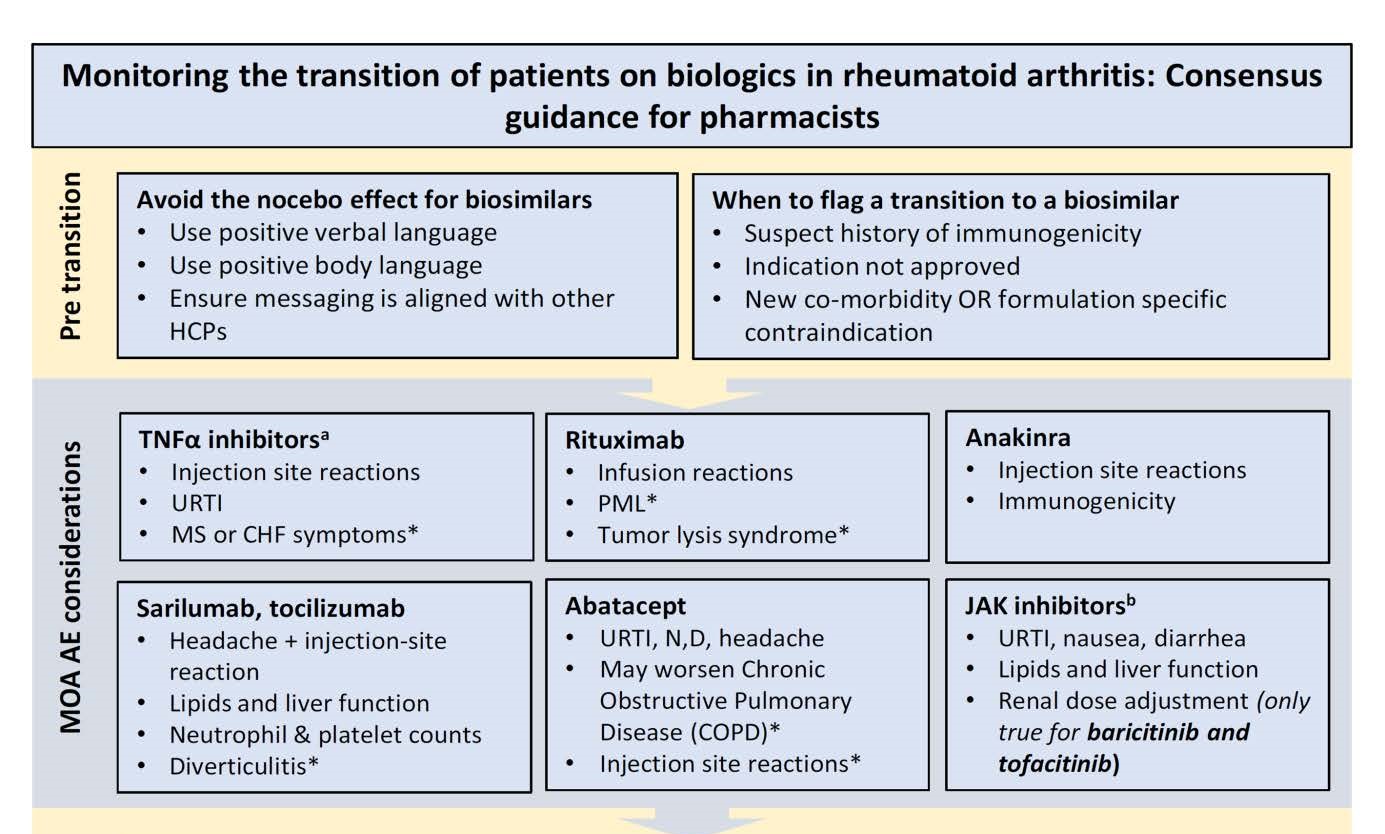
Preventing the nocebo effect requires a demonstration of confidence in biosimilars from all health care practitioners, including pharmacists, experts said in a newly published rheumatology treatment guidance document.
Rheumatology experts from Canada have developed pharmacist guidance for switching patients with rheumatologic disorders to biosimilars, which they hope also will be of use to the pharmacist community outside Canada.
The guidance was developed as Canadian provinces increasingly move toward mandatory switching to biosimilars to save money and improve access to care.
?New classes of antirheumatic drugs including janus kinase inhibitors (JAKs), along with the introduction of biosimilars are presenting more opportunity for therapeutic changes in patients with rheumatoid arthritis. These changes are often complex due to the nature of the drugs, along with an associated administrative burden,? they wrote. The group included physicians and pharmacists.
Consensus on Switching
In developing the guidance, the authors convened a panel of experts moderated by a facilitator and strived for consensus on key switching issues, primarily considerations prior to, during, and after transition to another biologic or JAK.
The panel paid close attention to the risks of the nocebo effect when switching therapies. Nocebo is when outcomes are affected by patient attitudes toward the therapy. The panel strongly recommended that pharmacists could best avoid nocebo by demonstrating confidence in the switch by using positive verbal language.
Consistent messaging from all involved health care providers was also seen as important to avoid this problem. ?The participants did not see the value of using a validated tool to screen and assess patient attitudes toward the therapeutic transition as a proxy for potential nocebo,? authors of the guidance wrote.
Although biosimilars are not precise copies of originator drug products, and even originator biologics differ from batch to batch, scientific evidence has shown that ?a single switch from a reference to a biosimilar is safe and effective. Additionally, clinical trials assessing switch from a bio-originator to a biosimilar have not demonstrated any loss of efficacy, increase in adverse events, or increased immunogenicity,? the authors wrote.
The group recommended that any concerns about potentially poor outcomes from a transition should be discussed with the prescribing rheumatologist prior to the switch.
Clinical ConsiderationsShown above is a portion of the clinical considerations tool developed by authors of the guidance.
The authors provided a list of clinical considerations pharmacists should be aware of during a therapeutic switch. These were based on the mechanism of action for each therapeutic category. For example, with the use of tumor necrosis factor?alpha inhibitors, pharmacists should be aware of injection site reactions, upper respiratory tract infection, or symptoms of congestive heart failure.
After the transition, the best tool available to assess disease activity is a visual analog scale for the assessment of pain, fatigue, and quality of life, the group agreed. Following close behind in the ranking of utility were the RAPID3 (Routine Assessment of Patient Index Data 3) tool, which is a disease activity index based on a questionnaire, and the Likert Scale, which measures patient agreement with various prewritten statements.
Pain, fatigue, and quality of life are helpful in assessing for primary response, which is whether the patient has any initial benefit from the agent. The pharmacist is best positioned to monitor for these outcomes, the authors wrote, because the patient will likely interact with the pharmacist for refills prior to a follow-up appointment with the rheumatologist.
?Although a lack of peak-response early in treatment is not a particularly concerning, a series of uncontrolled or frequent flare ups as compared to baseline after 3 months of therapy would be cause for immediate referral,? they wrote.
Asked when a pharmacist should refer a patient to the rheumatologist for an earlier-than-scheduled follow up, the group emphasized the seriousness and severity of individual factors such as infection, repeated flare ups, and surgery; it said these should be the basis of such determinations.
?The pharmacist is well positioned to monitor and resolve issues relating to adherence and safety,? the authors wrote.
In their clinical considerations tool, the authors provided a ?before starting? checklist to ensure a smooth transition to the substitute agent.
One of the questions considered by the panel but not addressed was how to measure the effectiveness of current therapy vs new therapy. ?Participants felt it was inappropriate to attempt this as it may create the impression that we expect a change in effectiveness post-transition. This could lead to a nocebo effect,? the authors wrote.
Reference
Choquette D, Chan J, Bardi M, et al. Monitoring the transition of patients on biologics in rheumatoid arthritis: consensus guidance for pharmacists.?Pharm Pract. 2021;19(3):2377. doi:10.18549/PharmPract.2021.3.2377
Studies of the infliximab biosimilar Remsima in patients with inflammatory bowel disease were presented at United European Gastroenterology Week 2021.
Investigators have reported positive findings from 2 studies of patients with Crohn disease (CD) or ulcerative colitis (UC) who switched from intravenous (IV) administration of the infliximab biosimilar Remsima (CT-P13) to subcutaneous (SC) administration.
Results from the inflammatory bowel disease (IBD) investigations were presented in poster form at United European Gastroenterology (UEG) Week 2021, being held virtually this week. Remsima is a Celltrion product.
In a randomized, controlled trial, investigators enrolled 65 patients (CD, 25; UC, 40) in the IV arm in which they received CT-P13 (5 mg/kg every 8 weeks from week 6 through week 22). At week 30, patients were switched to SC administration every 2 weeks until week 54 (dose 120 mg or 240 mg for patients < 80 kg or = 80 kg, respectively).
Investigators noted favorable outcomes in the SC group for pharmac
Share this article on WhatsApp, LinkedIn and Twitter